

Intracellular membrane trafficking fundamentally involves vesicles that bud from various membranes to be transported elsewhere within the endomembrane system of the eukaryotic cell. Our lab characterized the endosomal sorting complexes required for transport (ESCRTs) that induce a membrane budding process at the endosome that results in the formation of intralumenal vesicles (ILVs) inside the endosome, multivesicular endosome (MVE). This is a topologically inverted process from the formation of clathrin-coated endocytic vesicles, or COP-I and COP-II coated vesicles between the endoplasmic reticulum and the Golgi complex. A number of seemingly unrelated biological processes, including intralumenal vesicle formation inside multivesicular endosomes, enveloped virus budding at the plasma membrane (e.g., HIV), and the abscission event of metazoan cytokinesis, require the ESCRT machinery.
The ESCRTs comprise a pathway of five distinct complexes – ESCRTs-0, -I,-II, -III, and Vps4. Our long-term goal is to understand the molecular mechanisms that underlie the assembly and function of each of the ESCRT complexes in the formation of multivesicular endosomes for the delivery of cargo destined for degradation in the vacuole or lysosome. Previous studies in the lab set the framework establishing the ESCRT complexes as ubiquitin binding, cargo clustering and sequestering, and membrane remodeling machinery. Thus, the ESCRT pathway can be viewed as a cargo recognition and membrane deformation machine that can be analyzed from three distinct perspectives: 1) the membrane they deform, 2) the ESCRT proteins themselves, and 3) the cargoes they sort.
ESCRT-III has been shown to be the minimal machinery required for cargo sequestering and membrane invagination. Our current research interests focus on dissecting the protein-protein and protein-membrane interactions associated with the ESCRT-III complex. Using genetics, we found that ESCRT-III assembles transiently and dynamically on the endosomal membranes in a sequential order: Vps20-Snf7-Vps24-Vps2. The ESCRT-III complex is subsequently disassembled by the recruitment of the final ESCRT complex, the AAA-ATPase Vps4. By in vitro reconstitution assays using purified ESCRT-III components and model lipid systems, we described Snf7 filaments of ~9 nm that are converted into spiraling protein helices with the addition of Vps24 and Vps2. When coassembled with ESCRT-II, ESCRT-III forms ~65 nm diameter rings, indicative of a cargo-sequestering supercomplex. We have also recently identified two essential modules for ESCRT-III-membrane association. Our results indicate that ESCRT-III is tuned to maintain the topological constraints associated with protein filament-mediated membrane invagination and vesicle formation. Present work in the lab focuses on further investigating the conformational dynamics and molecular architecture of the ESCRT-III complex, through both in vivo genetic screening, and in vitro biochemical techniques.
Global Screening for Novel Endosomal Sorting Factors
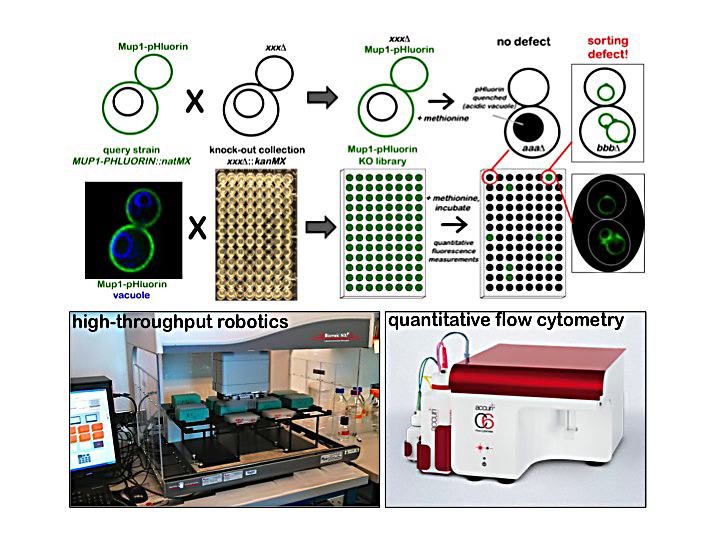
Cells need to constantly remodel the landscape of their surface to maintain cell integrity, growth, and homeostasis (e.g. ion channels, nutrient transporters, signaling receptors). In particular, cell surface growth factor receptors (e.g. EGFR) are precisely down-regulated through endocytosis and subsequent trafficking to the vacuole/lysosome for degradation. Failure to degrade these receptors can lead to aberrant cell signaling and cancer.
In the Emr lab, we have a long-standing interest in identifying novel genes/proteins necessary for trafficking transmembrane proteins through the endolysosomal membrane system. To uncover new components of endolysosomal trafficking, we developed a quantitative screening approach that utilizes the well-characterized cell surface transporter Mup1. Mup1 is the methionine permease of budding yeast, and exhibits rapid endocytic uptake when cells are exposed to exogenous methionine. We utilized Mup1-pHluorin, a fluorescent reporter that is trafficked to the acidic yeast vacuole, where its fluorescence is quenched by the acidity of the vacuole lumen. Using quantitative flow cytometry, we screened the yeast knock-out collection and identified mutants (including ESCRTs & ARTs) that exhibit substantial kinetic defects in the delivery of Mup1-pHluorin to the vacuole lumen. Future studies will dissect the roles for some of the novel proteins identified in the screen in endolysosomal trafficking.
Downregulation of receptor signaling is mediated by a series of membrane trafficking steps including ubiquitin-mediated endocytosis, ESCRT-mediated sorting and packaging into vesicles that bud into the lumen of the endosome, and endosome-lysosome fusion that delivers sorted receptors into the lumen of the lysosome where degradation occurs. Failure to attenuate plasma membrane (PM) signaling processes by endocytic downregulation can lead to hyperproliferation and cancer. To better understand endocytic downregulation, we are interested in deciphering how the ubiquitin conjugation machinery is targeted to specific proteins at the PM and how this targeting is regulated.
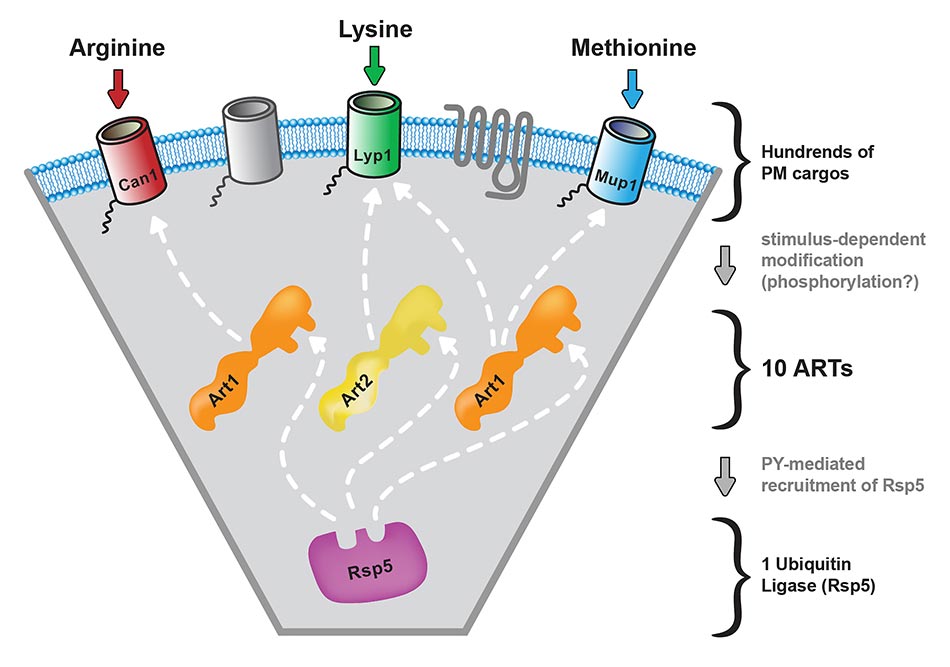
In Saccharomyces cerevisiae, ubiquitination mediated by Rsp5, the yeast Nedd4 homolog, is known to be required for the endocytic downregulation of numerous PM proteins. Previously, we identified and characterized a family of arrestin-related trafficking adaptors, or ARTs, which recruit the Rsp5 ubiquitin ligase to specific targets at the PM. Recent research from several groups has demonstrated that ART proteins are key determinants of target selection during ubiquitin-mediated endocytosis. Our goal is to understand how cargo selectivity and stimulus-dependent activation of ART proteins is achieved. We have found that Art1 undergoes a phosphoregulatory cycle and that dephosphorylation of Art 1 triggers its activation and recruitment to PM cargo. Using genetics, quantitative mass spectrometry and biochemistry, we have shown that this phosphoregulatory cycle is mediated by a TORC1-Npr1 kinase signaling cascade. In addition to phosphorylation/dephosphorylation, ARTs are also subject to ubiquitination. Present work focuses on dissecting the roles of deubiquitinases (DUB’s) in the localization, stability, activity and protein-protein interactions of Rsp5 and the ARTs.”
A key function of the ubiquitin-proteasome system is protein quality surveillance and the targeting of misfolded proteins for degradation. In the cytosol, specialized E3 ligases target soluble misfolded proteins for ubiquitination and subsequent proteasomal degradation. However, the case is more complicated for integral membrane proteins. Following co-translational insertion in the ER membrane, proteins that fail to fold properly in the ER are subject to ER-assisted degradation (ERAD), which involves retrotranslocation of proteins back into the cytosol followed by ubiquitin-dependent proteasomal degradation. Properly folded PM proteins, such as signaling receptors, ion channels, and nutrient transporters, exit the ER and traffic through the Golgi to the cell surface where they mediate their specific functions. Maintenance of proper PM proteostasis, particularly with respect to ion channels and nutrient transporters, is crucial to prevent loss of PM integrity and dissipation of essential ion and chemical gradients. As such, when PM resident proteins become damaged or misfolded, they must be recognized, removed by endocytosis and delivered to the lysosome for degradation. Thus, cells must maintain a “cradle to the grave” quality monitoring system for integral membrane proteins, yet the mechanisms of quality surveillance, particularly at the PM, remain poorly understood. Presently, we are analyzing the mechanism(s) by which misfolded proteins at the PM are detected and targeted for downregulation.
Phosphoinositide Lipid Signaling
{kind=link}
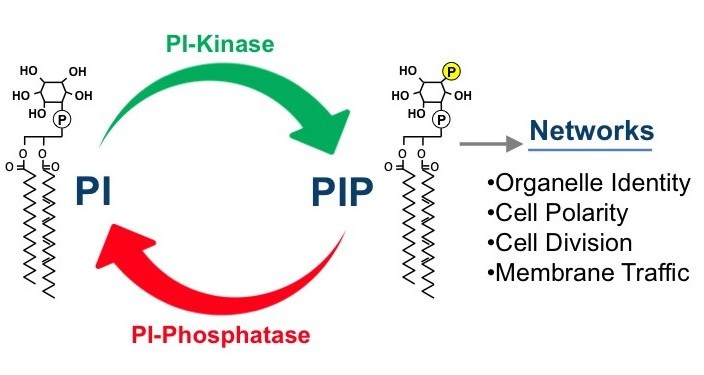
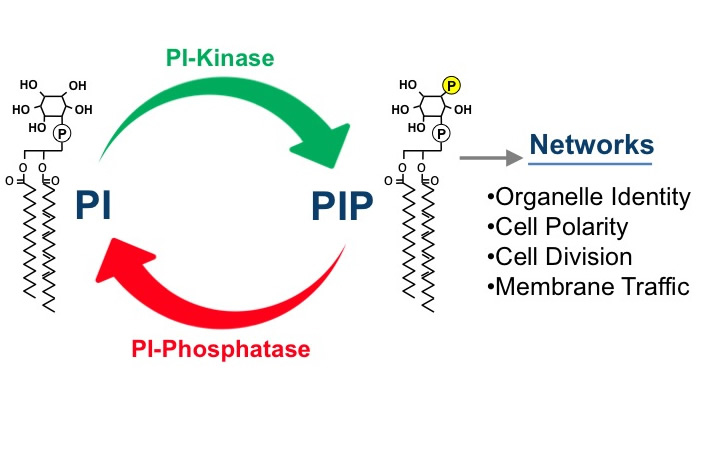
Phosphatidylinositol phosphate (PIP) lipids play an essential role in the regulation of diverse cellular processes, including cell growth, survival, differentiation, cytoskeletal organization, and membrane trafficking. Mutations in PIP metabolic genes are closely linked to serious human diseases including cancer. Specific PIP lipids are enriched in specific subcellular compartments, PI3P is enriched on membranes of the endosomal system, PI3,5P2 on the lysosome, PI4P on the Golgi (and PM) and PI4,5P2 on the plasma membrane. These compartment-specific PIP lipids provide a “lipid identity code” for each subcellular compartment by recruiting and activating specific effector proteins (containing domains such as PH, FYVE, PX, ENTH and GLUE domains) that directly bind the head group of the PI lipids. Our long-term goal has been to understand how the synthesis and turnover of PIPs are temporally and spatially regulated by lipid kinases and phosphatases using the powerful model organism yeast. In addition, we are analyzing the mechanisms by which each PIP regulates its distinct downstream cellular processes through interaction with specific effector proteins.
Synthesis and turnover of each PIP is regulated by a specific set of PIP kinases and phosphatases. Using yeast genetics and molecular biology techniques, and in vitro biochemical techniques, our research has contributed to understanding of the regulatory mechanisms of PIP metabolism. Our recent research includes discovery of unexpected regulatory pathways of PIP metabolism (PI4P and PI4,5P2) and PIP-dependent downstream cellular processes (PI3,5P2). We found that the PI4P phosphatase Sac1 is regulated by the Osh (OSBP) proteins. We also identified the ER-PM contact sites as the key regulatory sites for PI4P metabolism by Sac1. We also have shown that metabolic reprogramming from glycolysis to gluconeogenesis is regulated by PI3,5P2.
Our recent research has focused on the regulation and assembly of the PI 4-kinase and PI4P 5-kinase complexes at the PM, and the identification and function of novel PI3,5P2 effectors.
updated 7/18/14
